Stargardt Disease (STGD) is the most prevalent form of juvenile-onset macular dystrophy, predominantly due to gene mutations in an autosomal recessive inherited pattern. It is currently incurable as there are no Food and Drug Administration approved therapeutic treatments for STGD. The objective of “The Natural History of the Progression of Atrophy Secondary to Stargardt Disease (ProgStar)” studies is to determine the progression rate of STGD using a variety of measures.
WHAT IS STARGARDT DISEASE (STGD)?
Stargardt disease (STGD) is a progressive eye disease that compromises the function of the central part of the retina also known as the macula. The retina is a complex tissue lining in the inner surface of the back of the eye, which is responsible for converting light from the outside-world into a signal to the visual cortex in the brain. In people with STGD, the retinal pigment epithelium (RPE) that supports the light-sensitive photoreceptor cells in the macula fails to digest the metabolic waste from the photoreceptors, which leads to RPE and photoreceptor’s death and ultimately results in vision loss.
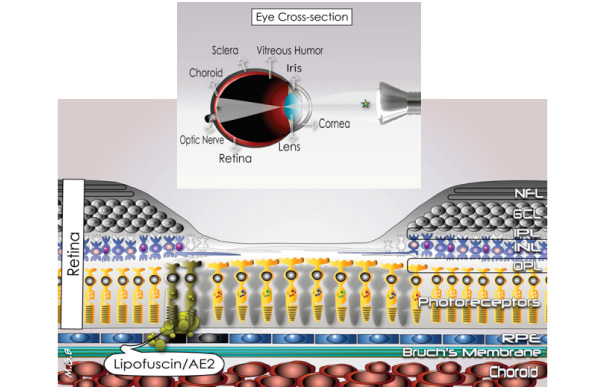
NFL: Nerve Fiber Layer; GCL: Ganglion Cells Layer; IPL: Inner Plexiform Layer; INL: Inner Nuclear Layer; OPL: Outer Plexiform Layer; RPE: Retinal Pigment Epithelium.
WHAT IS THE CAUSE OF STGD?
The disease is the most common juvenile macular degeneration with an estimated incidence of 10 – 12. 5 per 100,000 people [1]. Recently, it was shown that disease onset can be as late as in the 80s [2]. It is an inherited eye disease and in about 95% of cases is caused by mutations in the ABCA4 gene [1,3]. The ABCA4 gene is passed along to offspring in an autosomal recessive manner, meaning it has to be inherited from both parents in order for the disease to appear. Someone who inherits the gene from only one parent will be a carrier for STGD but will not have any symptoms. However, if that person has a child with someone else who is a carrier, there is a 25% probability that the child will have STGD. The carrier frequency for a mutation in the ABCA4 gene may be as high as 1:20 [4,5].
An autosomal dominant STGD-like phenotype [3] also has been described and is associated with mutations in the ELOVL4 genes [3,6,7]. In these cases, someone with only one copy of the mutation may manifest the disease.
STGD has been associated with considerable clinical and genetic heterogeneity. Mutations on at least three genes have been described and used to divide the disease into categories [11]:
- STGD1: The most common form of STGD. It is caused by mutations in the ABCA4 gene (short arm of chromosome 1p21-22), responsible for the codification of an adenosine triphosphate (ATP)-binding cassette transporter, which is localized to photoreceptors of the retina and is hypothesized to be involved in the clearance from photoreceptor cells of a byproduct of the retinoid (Vitamin A) cycle of vision [12-15]. Only about two thirds of patients with STGD1 have a detected mutation in this gene and over 600 disease-causing mutations in ABCA4 have been described [17]. Histologically the disorder is characterized by sub-retinal deposition of lipofuscin-like material.

- STGD3: There is also a rare dominant form caused by mutations in the ELOVL4 gene in chromosome 6q16. 6. Several different mutations in this gene have been described, including recessive mutation that will manifest with skin and brain dysfunction [18]. The gene codes a photoreceptor-specific membrane-bound protein that plays a role in long chain fatty acid biosynthesis. It is not known how the defective activity of the protein ELOVL-4 lead to lipofuscin accumulation.
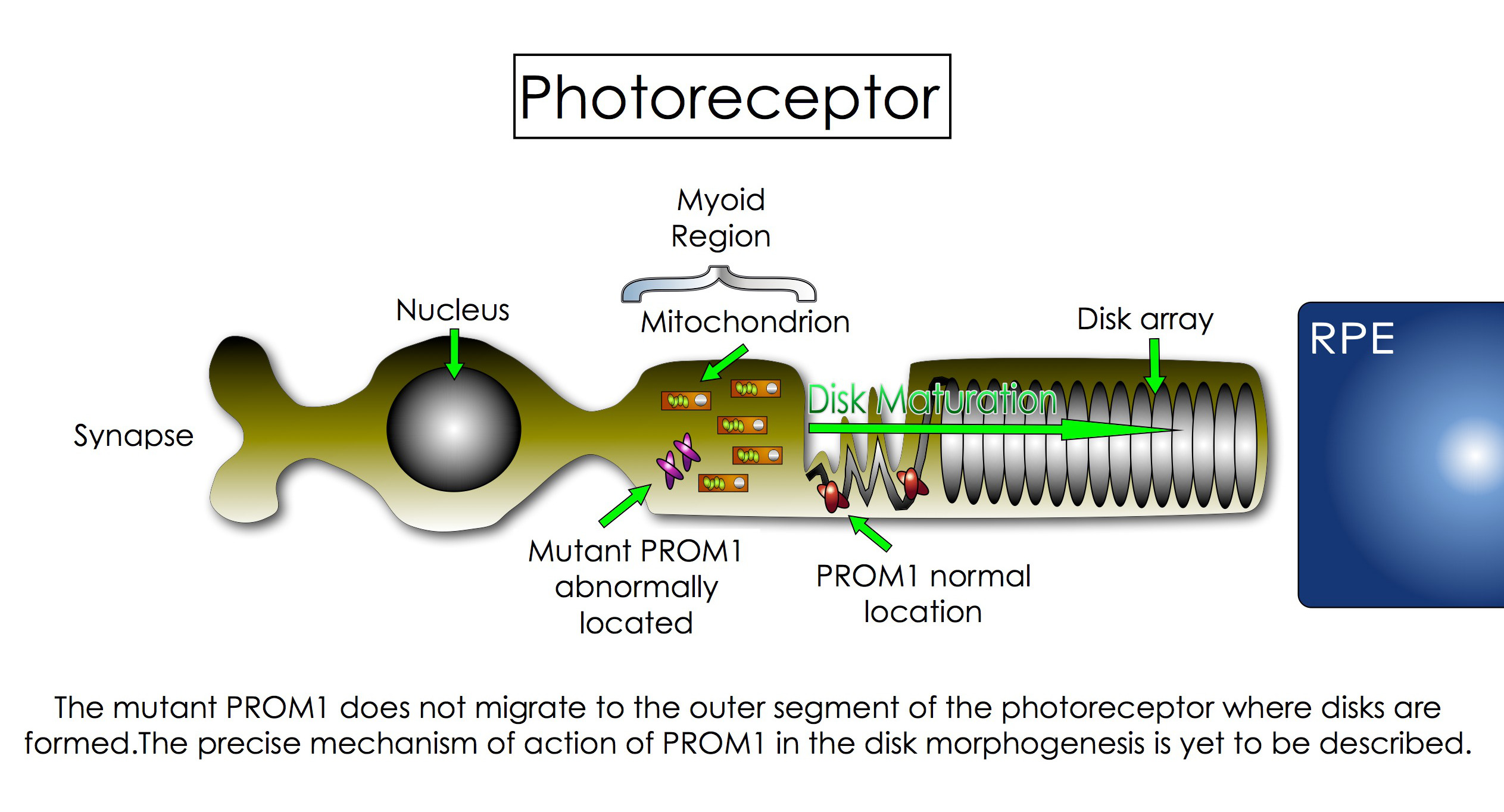
- STGD4: This is the most recent form of STGD to be described. The disease is caused by a mutation in the PROM1 gene mapped to chromosome 4 [25], which codes a protein called Prominin 1 (PROM1; also known as CD133 and AC133). PROM1 has an impact on the formation and organization of disks within the photoreceptors [26]. The disks are located in the outer segment (OS) of the cell and are the site where photochemical transformation of light occurs. In STGD4, the defective PROM1 is trapped in the myoid region of the photoreceptors and cannot migrate to the site where disks are formed. Ultimately, the absence of PROM1 in the OS affects the maturation of the disk, which leads to disk malfunction and to vision problems [27]. Although many advances in genetic science has helped to recognize this variant of STGD, some parts of this puzzle are yet to be discovered. For example, scientists still do not know how PROM1 interacts with the disks’ membrane and why its misallocation affects their maturation. More interestingly, scientists recently found that mutations on PROM1 gene could be transmitted as an autosome dominant trait and/or as an autosome recessive trait. The autosome dominant form is more associated with the STGD4 and other macular dystrophies, whereas the recessive trait is more related to cone-rod dystrophy and retinitis pigmentosa-like diseases [28].
Fundus flavimaculatus (FFM) is an allelic subtype of STGD and was shown to be caused by mutations in the chromosome 1p13 [23].
Despite the gene and mutation involved, a hallmark of STGD is premature accumulation of lipofuscin in RPE cells and in the sub-retinal space. Lipofuscin is a heterogeneous material derived from the chemically modified residues of incompletely digested photoreceptor outer segments. It is a heterogeneous material composed of a mixture of lipids, proteins, and different fluorescent compounds, the main fluorophore of which has recently been identified as a derivative of vitamin A, the di-retinal conjugate A2E and its photoisomers [8,20,24]. The AE2 photoisomers have toxic effect to cells, which can lead to cell death. The whole mechanism of lipofuscin formation and its role in disease pathogenesis are yet to be fully described.
Moreover, the marked phenotype (clinical appearance) variability, variable autosomal traits and large amount of mutations involving the ABCA4 gene make the correlation between gene mutations and phenotype a challenge for retina physicians.
WHAT ARE THE VISUAL SYMPTOMS?
It’s difficult to predict exactly when the disease will manifest and how fast it will progress. The course of the disease varies from person to person and members of the same family can show variations in course of disease.
The first manifestations can occur in childhood, but age of onset can even be in late in life. When onset occurs early, it usually has a more rapidly progressive course and poor final visual outcome.
Visual acuity can be severely reduced despite near normal peripheral visual fields. Central visual loss, loss of color vision, photophobia, paracentral scotoma, and slow dark adaptation are features of STGD.
The loss of acuity is accompanied by atrophic lesions in the macula and yellow-white pisciform deposits (referred to as “fundus flecks”). The deposits the retina are caused by excess accumulation of lipofuscin in the RPE. These may be present before clinical symptoms are present. Retinal vessel caliber is normal in STGD. Extensive macular disease can be associated with temporal pallor of the optic nerve. There are no systemic symptoms in STGD other than visual impairment.
HOW DO PHYSICIANS DIAGNOSE THE DISEASE?
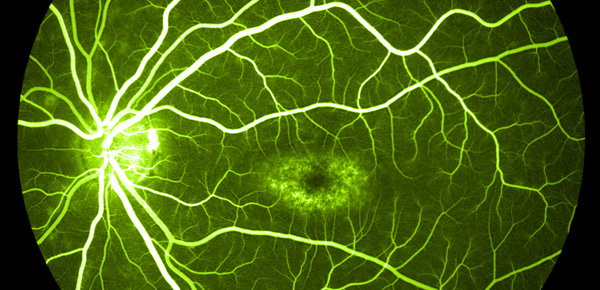
The diagnosis of STGD is mainly based on clinical characteristics including specific findings in fundus autofluorescence (FAF) imaging, optical coherence tomography (OCT) and fluorescein angiography (FA). Specifically, the natural autofluorescent properties of lipofuscin have led to the use of confocal scanning laser ophthalmoscopy (cSLO), and fundus autofluorescence imaging, as convenient, noninvasive methods for determining the distribution of lipofuscin in human participants.
More recently other diagnostic tools were incorporated in the assessment of STGD. Among them are screening of gene mutations (genotyping) and novel retinal imaging methods:
Common Genotyping Method

PREVENTION & THERAPIES
While there is no known treatment for STGD at this time, people with the condition often are recommended to protect their eyes against bright sunlight by wearing sunglasses and are advised not to supplement vitamin A or beta-carotene. There is no known treatment for STGD at this time.
CURRENT CLINICAL TRIALS
Ongoing Trials in US with or without participating sites outside US (as of 30 August 2013) / clinicaltrials. gov (click to enlarge).

References
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A et al: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997, 15(3):236-246.
- Fritsche LG, Fleckenstein M, Fiebig BS, Schmitz-Valckenberg S, Bindewald-Wittich A, Keilhauer CN, Renner AB, Mackensen F, Mossner A, Pauleikhoff D et al: A subgroup of age-related macular degeneration is associated with mono-allelic sequence variants in the ABCA4 gene. Invest Ophthalmol Vis Sci 2012, 53(4):2112-2118.
- Zhang K, Kniazeva M, Han M, Li W, Yu Z, Yang Z, Li Y, Metzker ML, Allikmets R, Zack DJ et al: A 5-bp deletion in ELOVL4 is associated with two related forms of autosomal dominant macular dystrophy. Nat Genet 2001, 27(1):89-93.
- Yatsenko AN, Shroyer NF, Lewis RA, Lupski JR: Late-onset Stargardt disease is associated with missense mutations that map outside known functional regions of ABCR (ABCA4). Hum Genet 2001, 108(4):346-355.
- Jaakson K, Zernant J, Kulm M, Hutchinson A, Tonisson N, Glavac D, Ravnik-Glavac M, Hawlina M, Meltzer MR, Caruso RC et al: Genotyping microarray (gene chip) for the ABCR (ABCA4) gene. Hum Mutat 2003, 22(5):395-403.
- Walia S, Fishman GA: Natural history of phenotypic changes in Stargardt macular dystrophy. Ophthalmic Genet 2009, 30(2):63-68.
- Walia S, Fishman GA, Kapur R: Flecked-retina syndromes. Ophthalmic Genet 2009, 30(2):69-75.
- Ma L, Kaufman Y, Zhang J, Washington I: C20-D3-vitamin A slows lipofuscin accumulation and electrophysiological retinal degeneration in a mouse model of Stargardt disease. J Biol Chem 2011, 286(10):7966-7974.
- Aveldano MI, Sprecher H: Very long chain (C24 to C36) polyenoic fatty acids of the n-3 and n-6 series in dipolyunsaturated phosphatidylcholines from bovine retina. J Biol Chem 1987, 262(3):1180-1186.
- Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J: Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc Natl Acad Sci USA 1998, 95(25):14609-14613.
- OMIM
- Sun H, Nathans J: ABCR: rod photoreceptor-specific ABC transporter responsible for Stargardt disease. Methods Enzymol 2000, 315:879-897.
- Cideciyan AV, Swider M, Aleman TS, Sumaroka A, Schwartz SB, Roman MI, Milam AH, Bennett J, Stone EM, Jacobson SG: ABCA4-associated retinal degenerations spare structure and function of the human parapapillary retina. Invest Ophthalmol Vis Sci 2005, 46(12):4739-4746.
- Kaufman Y, Ma L, Washington I: Deuterium enrichment of vitamin A at the C20 position slows the formation of detrimental vitamin A dimers in wild-type rodents. J Biol Chem 2011, 286(10):7958-7965.
- Molday RS, Molday LL: Identification and characterization of multiple forms of rhodopsin and minor proteins in frog and bovine rod outer segment disc membranes. Electrophoresis, lectin labeling, and proteolysis studies. J Biol Chem 1979, 254(11):4653-4660.
- Aveldano MI: Phospholipid species containing long and very long polyenoic fatty acids remain with rhodopsin after hexane extraction of photoreceptor membranes. Biochemistry 1988, 27(4):1229-1239.
- Burke TR, Tsang SH: Allelic and phenotypic heterogeneity in ABCA4 mutations. Ophthalmic Genet 2011, 32(3):165-174.
- Mandal MN, Ambasudhan R, Wong PW, Gage PJ, Sieving PA, Ayyagari R: Characterization of mouse orthologue of ELOVL4: genomic organization and spatial and temporal expression. Genomics 2004, 83(4):626-635.
- Bertolini G, Roz L, Perego P, Tortoreto M, Fontanella E, Gatti L, Pratesi G, Fabbri A, Andriani F, Tinelli S et al: Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci USA 2009, 106(38):16281-16286.
- Sparrow JR, Fishkin N, Zhou J, Cai B, Jang YP, Krane S, Itagaki Y, Nakanishi K: A2E, a byproduct of the visual cycle. Vision Res 2003, 43(28):2983-2990.
- Mata NL, Weng J, Travis GH: Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc Natl Acad Sci USA 2000, 97(13):7154-7159.
- Lewis RA, Lupski JR: Macular degeneration: the emerging genetics. Hosp Pract (1995) 2000, 35(6):41-50, 56-48.
- Gerber S, Rozet JM, Bonneau D, Souied E, Camuzat A, Dufier JL, Amalric P, Weissenbach J, Munnich A, Kaplan J: A gene for late-onset fundus flavimaculatus with macular dystrophy maps to chromosome 1p13. Am J Hum Genet 1995, 56(2):396-399.
- Saari JC, Garwin GG, Van Hooser JP, Palczewski K: Reduction of all-trans-retinal limits regeneration of visual pigment in mice. Vision Res 1998, 38(10):1325-1333.
- Kniazeva M, Chiang MF, Morgan B, Anduze AL, Zack DJ, Han M, Zhang K. A new locus for autosomal dominant stargardt-like disease maps to chromosome 4. Am J Hum Genet 1999, May, 64(5):1394-1399.
- Yang Z, Chen Y, Lillo C, Chien J, Yu Z, Michaelides M, et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J Clin Invest 2008, Aug, 118(8):2908-2916.
- Kleinman ME, Ambati J. Fifty years later: the disk goes to prom. J Clin Invest 2008, Aug, 118(8): 2681-2684.
- Michaelides M, Gaillard MC, Escher P, Tiab L, Bedell M, Borruat FX, et al. The PROM1 mutation p.R373C causes an autosomal dominant bull’s eye maculopathy associated with rod, rod-cone, and macular dystrophy. Invest Ophthalmol Vis Sci, 2010, Sep, 51(9):4771-4780.